
Abstract
Protein-ligand interactions are central to most biological processes. Understanding how proteins recognize ligands is crucial in biochemistry and molecular biology. Binding sites within proteins often form pockets that facilitate this recognition. However, identifying the specific residues within these sites that are critical for ligand binding can be challenging. This article introduces Alanine Binding Site-Scan (ABS-Scan), a freely accessible web server designed to systematically evaluate the contribution of individual binding site residues to ligand recognition through in silico alanine-scanning mutagenesis. ABS-Scan computationally mutates each residue in the binding site to alanine, calculates the ligand interaction energy for each mutant, and compares it to the wild-type protein to determine the ΔΔG values. This analysis provides a ranked list of residues based on their contribution to ligand interaction, enabling researchers to pinpoint potential loss-of-function mutations. The ABS-Scan web tool is available at: http://proline.biochem.iisc.ernet.in/abscan/.
Introduction
As of 2014, the Protein Data Bank (PDB) contained over 72,000 experimentally determined protein structures complexed with small molecule ligands 1, offering a wealth of data on protein binding sites. These sites vary in size, typically comprising six to thirty residues, depending on the ligand. However, the contribution of each amino acid to ligand binding is often unclear. Site-directed mutagenesis is a well-established experimental technique for studying the importance of specific residues 2. Alanine scanning mutagenesis (ASM) 3 takes this further by systematically mutating each residue to alanine to assess its functional impact. ASM has been successfully applied to study protein folding and stability 4, protein-protein interactions 5, 6, and protein-ligand interactions 7. Advances in high-throughput and cost-effective methods 8 have broadened the application of ASM. Yet, experimental biochemistry remains time-consuming and resource-intensive, leaving many proteins unexplored by ASM.
Computational alanine scanning mutagenesis has become increasingly viable due to the advancement of structural bioinformatics tools 9. The success of experimental ASM has spurred the development of in silico alanine scanning resources, including software like Modeller 10 and Rosetta 11. However, many of these tools are command-line based, posing accessibility challenges for some researchers. User-friendly web servers like Robetta 12, Rosetta Design 13, ROSIE 14, FOLDX 15, BeATMuSiC 16, and DrugScore PPI 17 cater to protein folding, stability, and protein-protein interaction studies. While computational workflows exist for evaluating ligand-binding energetics through methods like MM-GBSA 18– 20, a real-time, intuitive web tool for alanine scanning of small-molecule binding sites has been lacking. Experimental biochemists frequently need to identify key residues for mutagenesis to create loss-of-function mutants. A web-based tool addressing this specific need would be invaluable. Such a tool could also provide insights into critical interaction residues, identify residue sets that abolish ligand binding upon mutation, and aid lead refinement in drug discovery and understanding drug resistance mechanisms.
Here, we introduce ABS-Scan, a computational workflow and web server for automated alanine-scanning mutagenesis of protein-ligand interface residues. This alanine scanning tool integrates Modeller [10](#ref-10] for site-specific mutagenesis and Autodock [21](#ref-21] for protein-ligand complex energetic evaluation.
ABS-Scan Workflow: A Step-by-Step Guide to Alanine Scanning
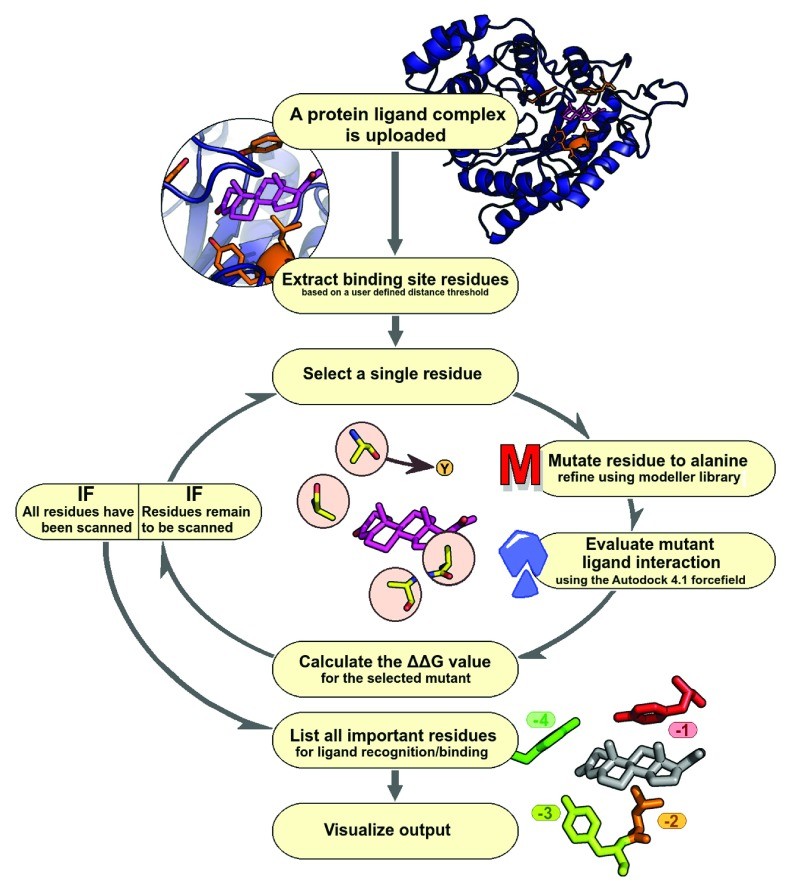
The ABS-Scan workflow is designed for user-friendliness and efficiency in alanine scanning analysis ( Figure 1). Users begin by submitting a protein-ligand complex of interest (PDB format). They can define the binding site by selecting a distance cutoff around the ligand. ABS-Scan then utilizes the Modeller library to perform site-specific mutagenesis, replacing each selected residue with alanine, followed by energy minimization 22. This minimization process involves conjugate gradient steps (200 iterations, minimum atom shift 0.001Å) and molecular dynamics simulation (200 steps) with steepest descent at varying temperatures. Restraints for the mutated models are derived from the wild-type protein structure.
The alanine scanning mutagenesis analysis relies on two key assumptions: (a) point mutations do not drastically alter the protein structure, and (b) ligand interaction modes remain consistent between mutant and wild-type complexes. Steric clashes are carefully avoided during minimization. The quality of generated mutant protein structures is assessed using the Discrete Optimized Protein Energy (DOPE) score 23, a statistical potential reflecting interaction feasibility and model compactness.
Figure 1. ABS-Scan workflow. Flowchart depicting various steps involved in ABS-Scan.
Each mutated structure is then scored using the Autodock 4.1 force field 21 to evaluate protein-ligand complex energetics. The force-field is used solely for scoring the protein-ligand interaction pose, without performing docking. Default settings include ‘check_hydrogens’ being ‘on’ and Gasteiger charges for proteins and ligands. The contribution of each residue is quantified as the difference in interaction score between the mutant and wild-type protein (∆∆G value). Results are presented graphically and as a ranked residue list, prioritizing residues for experimental site-directed mutagenesis. A Jmol applet visualizes protein-ligand interactions, coloring residues by their contribution, while a table displays intermolecular energy scores. A help section provides result explanations and examples.
Validation and Case Studies: Assessing ABS-Scan Performance
To validate the significance of the ∆∆G score in assessing residue contribution, ABS-Scan was tested on two datasets. The first dataset, from the CSAR Community Structure-Activity Resource (CSAR – www.csardock.org/), comprised native and artificial decoy complexes. ABS-Scan was successfully applied to 288 of 343 complexes. Decoy complexes, designed with ligands chemically similar to native ligands but known not to interact, showed a distinct average ∆∆G score distribution compared to native complexes ( Figure 2A & B). Native complexes had an average ∆∆G score of 0.395. The second dataset, derived from the PDBbind database 24 (195 protein-ligand complexes, PDBbind core dataset), yielded similar results. ABS-Scan successfully processed 135 complexes, with an average ∆∆G score of 0.387 per mutated residue. Based on these results, a stringent cutoff of 0.5 for ∆∆G score was chosen to indicate significant residue contribution. ABS-Scan effectively discriminated between decoy and native CSAR complexes in approximately 67% of cases (∆∆G ≥ 0.5; p-value ~0.004, Student’s t-test), indicating its ability to identify key residues for ligand interaction ( Figure 2C). Detailed ∆∆G scores for both datasets are available at http://proline.biochem.iisc.ernet.in/abscan/validation.
Figure 2. ABS-Scan Sensitivity. ( A) The average ∆∆G score per residue distribution from the cognate and decoy protein-ligand complexes of CSAR dataset. ( B) The scatter plot displaying the average ∆∆G score for native and the corresponding decoy complexes from the CSAR dataset. ( C) Boxplot showing the difference in the % of the residues in the binding site of cognate and decoy complexes having a predicted ∆∆G score ≥ 0.5.
Ideal validation would involve a dataset of binding affinities for wild-type and mutant proteins with the same ligand under uniform experimental conditions, which is scarce for protein-ligand interactions. While such datasets exist for protein-protein alanine scanning mutagenesis, none are readily available for protein-ligand systems. To compare ABS-Scan predictions with experimental alanine scanning data, a thorough literature search was conducted using the PDB and PubMed, yielding 126 structure hits and 56 citations. After filtering for biologically relevant ligands, metal ions, and modified residues, 79 entities/binding sites were identified (http://proline.biochem.iisc.ernet.in/abscan/validation). ABS-Scan was successfully applied to 54 of these structures, predicting at least two residues per binding site with ∆∆G score ≥ 0.5. The dataset and ranked residue lists are accessible at http://proline.biochem.iisc.ernet.in/abscan/validation.
Experimental alanine scanning studies report diverse mutant evaluation metrics (K d, K a, k cat/K M, etc.), making direct comparison challenging. Three case studies illustrate the heterogeneity of experimental data and ABS-Scan predictions:
Case Study 1: Rat 3-alpha-hydroxysteroid dehydrogenase (PDBID: 1AFS)
Heredia et al. 26 studied testosterone binding to rat 3-alpha-hydroxysteroid dehydrogenase, reporting Kd values for mutants. ABS-Scan analysis of the enzyme complexed with testosterone and progesterone predicted W227 (∆∆G score = 1.43, Kd = 10.7±1.2), Y310 (∆∆G score = 1.31, Kd = 9.20±0.94), and L54 (∆∆G score = 0.5696, Kd = 7.24±0.79) as important. A good correlation was observed between predicted ∆∆G scores and experimental Kd values (0.829 for testosterone, 0.704 for progesterone).
Case Study 2: Vitamin-D receptor (PDBID: 1IE9)
Shimizu et al. 27 performed alanine scanning on the vitamin-D receptor with vitamin-D analogs. ABS-Scan analysis of docked vitamin-D analogs (Rosetta 3.4) on the receptor identified L233, W286, R274, and H397 as crucial residues (∆∆G score > 0.5) for interaction with all four analogs. These residues, involved in hydrophobic and hydrogen bond interactions, aligned with experimental transactivation assay results.
Case Study 3: Human trimethyl-guanosine synthase (Tgs1) (PDBID: 3GDH)
A study on human Tgs1 by Gonatopoulos-Pournatzis et al. 29 investigated interactions with mGTP and AdoMet. ABS-Scan predicted R807 (∆∆G score 3.63) and K646 (∆∆G score 3.39) as essential, consistent with experimental methyltransferase assay results. W766 (∆∆G score 2.66), involved in π -cation stacking, was also identified as crucial, correlating with the absence of methylated products in mutants.
Details of these case studies and analysis results are available in the examples section of the web tool: http://proline.biochem.iisc.ernet.in/abscan/examples. These case studies demonstrate the utility of Alanine Scanning Tools like ABS-Scan in predicting functionally important residues.
Implementation: Building the ABS-Scan Web Server
The ABS-Scan web server is built using hypertext preprocessor (PHP). It integrates libraries from Autodock, Modeller, and Pymol for mutation modeling and energy evaluation. Shell, Python, Java, HTML, and PHP scripts facilitate the integration of these back-end libraries into a user-friendly interface. The server is platform-independent and accessible via any web browser. For advanced users, a command-line interface is available as a Python script on GitHub (https://github.com/praveeniisc/ABS-Scan). The script is compatible with Linux OS (Ubuntu 12.04) and requires Modeller 10, MGL AutodockTools 30, and Pymol (http://pymol.org). The web server utilizes the d3.js library for plotting and Jmol Applet for visualizing protein-ligand interactions.
Input: Setting Up Your Alanine Scan
ABS-Scan requires a protein-ligand complex structure in PDB format as input. Users can provide a four-letter PDB ID or upload a PDB file. The server allows users to define a distance cutoff and select the ligand to specify binding site residues for alanine scanning. A default 4.5 Å cutoff is set to select residues within this distance from any ligand atom.
Figure 3. ABS-Scan interactive display. Snapshot explaining the Jmol applet output on the ABScan server. The individual residues are colored in red to blue gradient depending upon the contribution towards the ligand interaction as predicted by ABScan ∆∆G score. Options to visualize the different kinds of interaction – polar, hbonds etc. is also provided.
For cases involving metal ions 31 or water molecules 32 crucial for interactions, an advanced option allows users to upload a PDBQT format ligand file. This accommodates unusual atom types, metal ions, or bridge-water molecules. For instance, bridge-water molecules can be included in the ligand PDBQT file. ABS-Scan analysis of protein lysine methyltransferase (PDB: 3S7B) with S-adenosyl methionine 33 and bridge-water molecules successfully identified GLU135 and ASN182 as significant contributors through water bridges, demonstrating the tool’s advanced capabilities.
Output: Interpreting Alanine Scanning Results
ABS-Scan results are interactively visualized on the web server. The Jmol Applet ( Figure 3) displays residue contributions to ligand interaction, color-coded according to ∆∆G scores.
Figure 4. ABS-Scan energy plots. ( A) ∆∆G values reported for each of the alanine mutation performed for the residues present at the binding site. The residues are ordered according to their contribution/∆∆G values. ( B) The different energy component of autodock interaction score plotted for each of the alanine mutant produced at the binding site.
The d3.js library generates plots of predicted ∆∆G values and Autodock4 energy components ( Figure 4). Users can download publication-quality images (SVG/PDF/PNG), raw mutant PDB files, and CSV files containing ∆∆G and Autodock energy scores. The Twitter Bootstrap Java library provides web server framework development.
Conclusions
The ABS-Scan web server is a valuable alanine scanning tool for gaining insights into protein-ligand molecular recognition. It allows researchers to analyze experimentally determined protein-ligand structures and modeled complexes to understand residue-level contributions to ligand binding. ABS-Scan enhances binding site analysis, facilitates comparison of ligand interactions, and is a significant resource for ASM studies.
Software Availability
Software access
http://proline.biochem.iisc.ernet.in/abscan/
Latest source code
https://github.com/praveeniisc/ABS-Scan
Source code as at the time of publication
https://github.com/F1000Research/ABS-Scan/releases/tag/V1.0
Archived source code as at the time of publication
http://dx.doi.org/10.5281/zenodo.12806 34
Software license
ABS-Scan is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Acknowledgements
We acknowledge all members of the NSC lab for their valuable suggestions during the development of the web server and result visualization.
Funding Statement
The authors declare no specific grants were sanctioned for this project. PA was supported by a Bristol-Myers Squibb fellowship during this work.
References
[1] (ref-1) F1000Res. 2016 Jan 19. doi: 10.5256/f1000research.6207.r11974
[2] (ref-2) F1000Res. 2015 Feb 4. doi: 10.5256/f1000research.6207.r7324
[3] (ref-3) F1000Res. 2014 Dec 22. doi: 10.5256/f1000research.6207.r7122
[4] (ref-4) F1000Res. 2014 Oct 17. doi: 10.5256/f1000research.5509.r6276
[5] (ref-5) Bernard Offmann. Referee response for version 2. F1000Res. 2016 Jan 19. doi: 10.5256/f1000research.6207.r11974
[6] (ref-6) Srikrishna Subramanian, Gurmeet Kaur. Referee response for version 2. F1000Res. 2015 Feb 4. doi: 10.5256/f1000research.6207.r7324
[7] (ref-7) Sunando Datta. Referee response for version 2. F1000Res. 2014 Dec 22. doi: 10.5256/f1000research.6207.r7122
[8] (ref-8) Bernard Offmann, Stéphane Téletchéa. Referee response for version 1. F1000Res. 2014 Oct 17. doi: 10.5256/f1000research.5509.r6276
[9] (ref-9) Nagasuma Chandra. F1000Res. 2014 Nov 18.
[10] (ref-10) Šali A, Blundell TL: Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993 Dec 5;234(3):779-815. PubMed PMID: 8254808
[11] (ref-11) Leaver-Fay A, Tyka M, Lewis SM, et al.: ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545-74. PubMed PMID: 21073985
[12] (ref-12) Kim DE, Chivian D, Baker D: Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W526-31. PubMed PMID: 15215425
[13] (ref-13) Lyskov S, Chou FC, Conchúir SÓ, et al.: Server for protein design. Proteins. 2013 Dec;81(12):2174-82. PubMed PMID: 23996729
[14] (ref-14) Conchúir SÓ, Lyskov S, Ashworth J, et al.: ROSIE: a web server for protein structure prediction and design. Bioinformatics. 2010 Nov 1;26(21):2839-40. PubMed PMID: 20829399
[15] (ref-15) Guerois R, Nielsen JE, Serrano L: Predicting changes in protein stability upon mutation using the FoldX force field. J Mol Biol. 2002 Dec 6;320(2):369-80. PubMed PMID: 12095135
[16] (ref-16) Dehouck Y, Grosjean H, Gilis V: BeAtMuSiC: prediction of changes in protein stability and melting temperature following point mutations. Bioinformatics. 2011 Jun 15;27(11):1527-8. PubMed PMID: 21474513
[17] (ref-17) Günther G, Steinmetzer T, Sotriffer CA: DrugScorePPI web server: fast and accurate prediction of protein-protein interaction hot spots. Nucleic Acids Res. 2011 Jul;39(Web Server issue):W270-5. PubMed PMID: 21593140
[18] (ref-18) Kollman PA, Massova I, Reyes C, et al.: Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000 Dec;33(12):889-897. PubMed PMID: 11124044
[19] (ref-19) Srinivasan J, Wang C, Kollman PA, Case DA: Continuum solvent studies of protein structure and thermodynamics. J Am Chem Soc. 1998 Oct 21;120(41):10525-10530. PubMed PMID: 11670515
[20] (ref-20) Tsui V, Case DA: Theory and applications of the generalized Born/surface area model in macromolecular simulations. Biopolymers. 2001;56(4):275-91. PubMed PMID: 11753995
[21] (ref-21) Morris GM, Huey R, Lindstrom W, et al.: AutoDock4 and AutoDockTools4: Automated docking suite. J Comput Chem. 2009 Nov;30(16):2785-91. PubMed PMID: 19399780
[22] (ref-22) van Gunsteren WF, Billeter SR, Eising AA, et al.: Biomolecular Simulation: The GROMOS96 Manual and User Guide. Zürich: Vdf Hochschulverlag AG an der ETH Zürich; 1996.
[23] (ref-23) Shen MY, Sali A: Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006 Nov;15(11):2507-24. PubMed PMID: 17075131
[24] (ref-24) Wang R, Fang X, Lu Y, Wang S: The PDBbind database: Collection of binding affinities for protein-ligand complexes with known 3D structures. J Med Chem. 2004 Aug 12;47(12):2977-80. PubMed PMID: 15161247
[25] (ref-25) Thorn KS, Bogan AA, McCallum EO, et al.: Combinatorial alanine scanning of an antibody CDR loop. J Mol Biol. 1997 Dec 5;273(5):1128-42. PubMed PMID: 9398484
[26] (ref-26) Heredia VV, Werck-Reichhart D, Poirey R, et al.: Importance of the active site residue tryptophan 227 for catalytic activity of rat liver 3 alpha-hydroxysteroid dehydrogenase. Biochemistry. 1996 Jul 23;35(29):9521-8. PubMed PMID: 8692485
[27] (ref-27) Shimizu C, Pike JW, Norman AW, et al.: Two-dimensional alanine-scanning mutagenesis of the vitamin D receptor hormone binding domain reveals residues critical for ligand-induced transactivation. J Biol Chem. 1997 Nov 28;272(48):30749-58. PubMed PMID: 9374508
[28] (ref-28) Chaudhury S, Lyskov S, Gray JJ: PyRosetta: a Python-interfaced molecular modeling suite for de novo design and structural bioinformatics. Bioinformatics. 2010 Jun 1;26(10):1292-9. PubMed PMID: 20385755
[29] (ref-29) Gonatopoulos-Pournatzis T, Cowling VH, Cole MD, et al.: Human TGS1/PIMT is a trimethylguanosine synthase required for snRNA and snoRNA biogenesis. Mol Cell. 2004 Nov 19;16(4):547-59. PubMed PMID: 15541446
[30] (ref-30) Sanner MF: Python: a programming language for software integration and development. J Mol Graph Model. 1999 Jun;17(1):57-61. PubMed PMID: 10381722
[31] (ref-31) Dudev T, Lim C: Metal-ligand interactions in proteins. Chem Rev. 2003 Jan;103(1):2167-202. PubMed PMID: 12528998
[32] (ref-32) Levy Y, Onuchic JN: Water mediation in protein folding and molecular recognition. Annu Rev Biophys Biomol Struct. 2006;35:389-415. PubMed PMID: 16649892
[33] (ref-33) Zhang X, Zeng L, Yang J, et al.: Structure of the SET7 domain-MLL1 core complex reveals the basis for site-specific lysine methylation. Mol Cell. 2009 Jul 24;35(2):277-87. PubMed PMID: 19635559
[34] (ref-34) Praveen Kumar A, Chandra N: Archived source code as at the time of publication. http://dx.doi.org/10.5281/zenodo.12806. 2014.