Introduction
Protein–ligand interactions are fundamental to most biological processes within living organisms. The intricate dance between proteins and ligands dictates a vast array of physiological functions. A crucial aspect of understanding these processes lies in deciphering how proteins recognize and bind to their specific ligands. Protein binding sites, often forming pockets within the protein structure, are responsible for creating the precise environment needed for ligand recognition. However, the exact boundaries and key residues within these binding sites are frequently not well-defined.
To address this challenge, alanine scanning mutagenesis has emerged as a powerful technique. This method systematically assesses the importance of individual amino acids within a protein’s binding site. Traditionally, alanine scanning mutagenesis involves the experimental creation of point mutants, where each residue of interest is mutated to alanine, and the effect of this mutation on protein function is then evaluated. This experimental approach, while valuable, is often time-consuming, costly, and labor-intensive, limiting its application to a broad range of proteins.
The advent of structural bioinformatics tools has paved the way for computational, or in silico, alanine scanning mutagenesis. Several computational resources now exist that leverage the principles of alanine scanning, including well-known software packages like Modeller and the Rosetta suite. However, many of these resources are command-line based, posing a barrier to entry for researchers without strong computational skills. While web servers exist for protein folding, stability, and protein-protein interactions, a user-friendly, real-time web tool specifically designed for analyzing alanine-scanning mutations in small-molecule binding sites has been lacking.
Recognizing this gap, we developed Alanine Binding Site-Scan (ABS-Scan), a computational workflow and web server designed for automated in silico alanine-scanning mutagenesis of protein-ligand interface residues. ABS-Scan aims to provide an intuitive and accessible Alanine Scanning Tool to the research community. This web server empowers experimental biochemists to efficiently identify critical amino acids for ligand binding, facilitating the generation of loss-of-function mutants. Beyond mutant generation, ABS-Scan offers deeper insights into crucial interaction residues, residue pairs, and sets that are vital for ligand binding. This analytical capability is invaluable for lead refinement in drug discovery and understanding mechanisms of drug resistance arising from mutations.
The ABS-Scan workflow integrates established software libraries, including Modeller for site-specific alanine mutagenesis and Autodock for evaluating protein-ligand complex energetics. This combination provides a robust and efficient platform for alanine scanning analysis. The web server, freely accessible at http://proline.biochem.iisc.ernet.in/abscan/, offers researchers a valuable alanine scanning tool to advance their understanding of protein-ligand interactions.
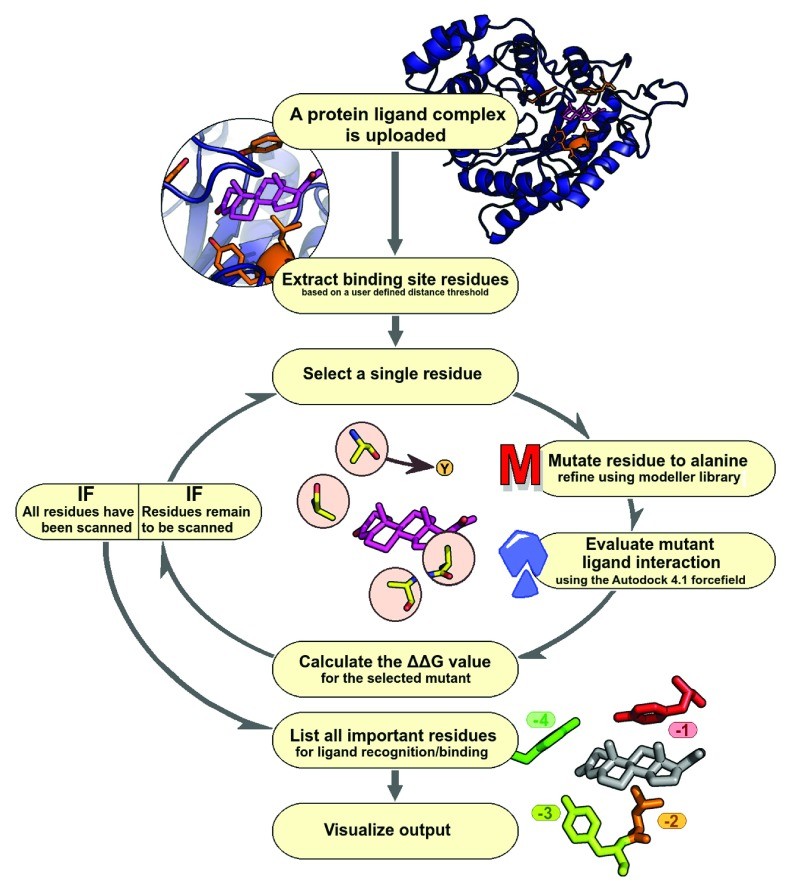
{width=790 height=893}Workflow of the Alanine Scanning Tool
The ABS-Scan workflow is designed to be user-friendly and efficient, allowing researchers to easily analyze protein-ligand complexes (Figure 1). Users begin by submitting the structure of their protein-ligand complex, either by providing a four-letter PDB ID or uploading a PDB structure file. A key feature of this alanine scanning tool is the option to define the binding site. Users can select a distance cut-off to specify the region around the ligand that will be subjected to in silico alanine scanning mutagenesis. This allows for focused analysis of residues directly involved in ligand interaction.
Once the input parameters are set, the workflow utilizes the Modeller library to perform site-specific mutagenesis. Each selected residue within the defined binding site is computationally mutated to alanine. To ensure structural integrity after mutation, the workflow incorporates energy minimization steps. This process involves an initial conjugate gradient minimization, followed by molecular dynamics simulation with steepest descent at varying temperatures. Restraints for the mutated model are derived from the original wild-type protein structure, ensuring minimal structural perturbation from the mutation. The system is carefully minimized to prevent steric clashes between protein and ligand atoms.
To assess the quality of the generated mutant protein structures, ABS-Scan reports Discrete Optimized Protein Energy (DOPE) scores. The DOPE score is a statistical potential score that reflects the feasibility and compactness of the modeled structure, providing an indication of model quality after mutagenesis and minimization.
Following structural modeling, each mutated structure undergoes energetic evaluation using the Autodock 4.1 force field. This force field is employed to calculate the protein-ligand interaction energy for both the wild-type and mutant complexes. It’s important to note that Autodock is used here solely for scoring the existing protein-ligand pose, and no docking is performed. By default, the workflow prepares the receptor by turning ‘check_hydrogens’ flag ‘on’ and assigns Gasteiger charges to both protein and ligand, standardizing the energetic calculations.
The core output of the alanine scanning tool is the ΔΔG value, which represents the difference in interaction score between the mutant and wild-type protein. This value quantifies the contribution of each mutated residue to ligand interaction. ABS-Scan then presents these results to the user in an intuitive and informative manner. A ranked list of residues, based on their ΔΔG values, is provided, highlighting the residues predicted to be most critical for ligand binding. This ranked list directly assists in designing site-directed mutagenesis experiments to generate loss-of-function mutants. Furthermore, a Jmol applet dynamically visualizes protein-ligand interactions, coloring residues based on their contribution to the interaction as predicted by the ΔΔG score. Simultaneously, a table displays the intermolecular energy scores, providing detailed numerical data. Finally, a comprehensive help section is included within the web server, offering explanations of the results and illustrative examples to guide users in interpreting the output of this alanine scanning tool.
{width=770 height=624}Validation and Case Studies of the Alanine Scanning Tool
To rigorously evaluate the significance of the ΔΔG score and the overall performance of the ABS-Scan alanine scanning tool, we conducted a comprehensive validation study using two distinct datasets.
The first dataset was derived from the CSAR (Community Structure-Activity Resource) database. This dataset is particularly valuable for validation as it includes “decoy” complexes, which are artificially generated docked complexes of proteins with ligands that possess similar chemical properties to native ligands but are known not to interact with the protein. Our analysis successfully processed 288 out of 343 protein-ligand native and decoy complexes from the CSAR dataset. The distribution of average ΔΔG scores for binding site residues in decoy complexes was demonstrably different from that of native protein-ligand complexes (Figure 2A & B). Native complexes exhibited an average ΔΔG score of 0.395, indicating a stronger energetic contribution from binding site residues.
The second validation dataset was sourced from the PDBbind database, a widely recognized resource for protein-ligand binding affinity data. We utilized the PDBbind core dataset, comprising 195 protein-ligand complexes, and successfully processed 135 of these using the ABS-Scan workflow. In this dataset, the average ΔΔG score observed for each mutated residue at the binding site was 0.387, consistent with the findings from the CSAR native complex dataset.
Based on these validation studies, we established a stringent ΔΔG score cut-off of 0.5 to determine the sensitivity of ABS-Scan. Using this cut-off, ABS-Scan effectively discriminated between decoy and native complexes in the CSAR dataset in approximately 67% of cases (p-value ~0.004, Student’s t-test). This significant discrimination capability (Figure 2C) underscores the tool’s ability to reliably identify residues crucial for ligand interaction. Detailed results of ΔΔG scores for each mutation in both datasets are available on the web-resource: http://proline.biochem.iisc.ernet.in/abscan/validation.
While ideal validation would involve a comprehensive dataset of experimentally determined binding affinities for both wild-type and mutant proteins with the same ligand, such a dataset is not readily available for protein-ligand interactions. To further assess ABS-Scan’s predictive power against experimental data, we conducted an extensive literature search to identify published alanine-scanning mutagenesis studies of protein-ligand binding sites. Using advanced search options in PDB and scanning PubMed abstracts for “alanine scanning,” we curated a dataset of 79 entities/binding sites with experimental alanine scanning data. After filtering for biologically relevant ligands and excluding metal ions and modified residues, we successfully performed alanine scanning using ABS-Scan on 54 of these structures. On average, ABS-Scan predicted at least two residues per binding site to have a ΔΔG score ≥ 0.5, further supporting the tool’s sensitivity. The complete dataset and ranked residue lists are accessible at http://proline.biochem.iisc.ernet.in/abscan/validation, providing a valuable resource for the community.
To illustrate the application and heterogeneity of experimental validation measures, we present three case studies that compare ABS-Scan predictions with diverse experimentally reported mutant evaluation scores.
Case Study 1: Rat 3-alpha-hydroxysteroid dehydrogenase (PDBID: 1AFS)
Heredia et al. investigated the testosterone binding site of rat 3-alpha-hydroxysteroid dehydrogenase. Their study, using experimental alanine scanning, reported Kd values to assess binding affinity. ABS-Scan analysis of 3-alpha hydroxysteroid dehydrogenase in complex with testosterone and progesterone correctly identified residues W227 (ΔΔG score = 1.43; Kd = 10.7±1.2), Y310 (ΔΔG score = 1.31; Kd = 9.20±0.94), and L54 (ΔΔG score = 0.5696; Kd = 7.24±0.79) as important for ligand recognition. Notably, a good correlation was observed between the reported Kd values and the predicted ΔΔG scores (0.829 for testosterone and 0.704 for progesterone), demonstrating the tool’s ability to reflect experimental binding affinity changes.
Case Study 2: Vitamin-D receptor (PDBID: 1IE9)
Shimizu et al. performed two-dimensional alanine scanning mutations to study the structure-function relationship of the vitamin-D receptor and vitamin-D analogs. Using a transcriptional activity assay (luciferase activity) to evaluate mutant effects, they assessed the functional impact of mutations. ABS-Scan analysis, performed on docked poses of four vitamin-D analogs with the receptor, identified residues L233, W286, R274, and H397 as crucial for interaction with all analogs, exhibiting ΔΔG scores > 0.5. These residues, located in helix 3, β-sheet, and helix 4, are known to engage in hydrophobic and hydrogen bond interactions with the ligand, consistent with the experimental findings and demonstrating the tool’s capacity to predict functionally important residues.
Case Study 3: Human trimethyl-guanosine synthase enzyme (Tgs1) (PDBID: 3GDH)
A study on human trimethyl-guanosine synthase enzyme (Tgs1) by Gonczy et al. investigated interactions with m7GTP and AdoMet using methyltransferase assays to quantify methylation levels. ABS-Scan analysis correctly predicted residues R807 and K646, reported as the most affected mutants in the experimental study, to be essential, with the highest ΔΔG scores of 3.63 and 3.39, respectively. These positively charged residues interact with phosphate groups of m7GTP. Furthermore, ABS-Scan also highlighted W766 (ΔΔG score of 2.66), involved in π-cation stacking with m7G, as crucial, which aligned with experimental observations of abolished methylated products for this mutant.
These case studies, along with further examples, are available on the web-tool’s example section: http://proline.biochem.iisc.ernet.in/abscan/examples. They collectively demonstrate the ABS-Scan alanine scanning tool’s ability to provide valuable and experimentally relevant insights into protein-ligand interactions across diverse systems and experimental assays.
Implementation of the Alanine Scanning Web Server
The ABS-Scan web server is built upon a robust and flexible architecture, implemented using hypertext preprocessor (PHP). It seamlessly integrates established software libraries, including Autodock, Modeller, and Pymol, for mutation modeling and energetic evaluation. Shell, Python, Java, HTML, and PHP scripts facilitate the integration of these back-end libraries into a functional and user-friendly web interface. The server is designed to be platform-independent, ensuring accessibility from any machine with internet access and a web browser.
For advanced users who prefer command-line operation, a standalone python script is available through a github repository: https://github.com/praveeniisc/ABS-Scan. This script has been tested on a standard Intel 2.83 GHz quad-core system running 32-bit Linux OS (Ubuntu 12.04) with Modeller, MGL AutodockTools, and Pymol installed.
The web server utilizes the d3.js library for generating interactive and publication-quality plots of the results. Protein-ligand interaction visualization is achieved through a Jmol Applet, providing dynamic and insightful visual representations. The Twitter bootstrap java library provides the framework for the web server’s development, ensuring a responsive and modern user interface. Users have the option to download publication-quality images in SVG/PDF/PNG formats, as well as raw data files including individual mutant PDB structures, ΔΔG scores in CSV format, and Autodock energy scores, facilitating further analysis and integration with other research workflows.
Input to the Alanine Scanning Tool
The primary input for the ABS-Scan server is the structure of a protein-ligand complex in PDB format. Users can input this data either by providing the four-letter PDB ID or by uploading a PDB structure file directly. To define the binding site for alanine scanning, the tool offers a crucial option: users can specify a distance cut-off. By default, this cut-off is set to 4.5 Å, selecting all residues with atoms within this distance from any ligand atom. This distance parameter allows users to fine-tune the binding site definition based on their specific research question.
Recognizing the potential role of metal ions and water molecules in protein-ligand interactions, ABS-Scan provides advanced input options. While metal ions and water molecules can be critical for stabilizing interactions, incorporating them directly into the automated workflow poses challenges. For instance, metal ions can exist in various ionic states, each requiring specific charge parameters for accurate energetic calculations. Similarly, the reliable inclusion of structural water molecules depends heavily on the resolution of the crystal structure.
To address these complexities, ABS-Scan offers an advanced option that allows users to upload the ligand in PDBQT format. This option is particularly useful in cases where the ligand contains unusual atom types, metal ions, or involves bridge-water molecules in its interaction with the protein. For practical purposes, bridge water molecules can be considered as part of the ligand and incorporated into the ligand’s PDBQT file. As an example, the manuscript illustrates the use of this advanced option with protein lysine methyltransferase (PDB: 3S7B) complexed with S-adenosyl methionine, where four bridge water molecules are crucial for the interaction. By including these water molecules in the ligand PDBQT file, ABS-Scan correctly identified GLU135 and ASN182 as significant contributors to ligand binding via water bridges. This example highlights the flexibility and advanced capabilities of the ABS-Scan alanine scanning tool.
Output from the Alanine Scanning Tool
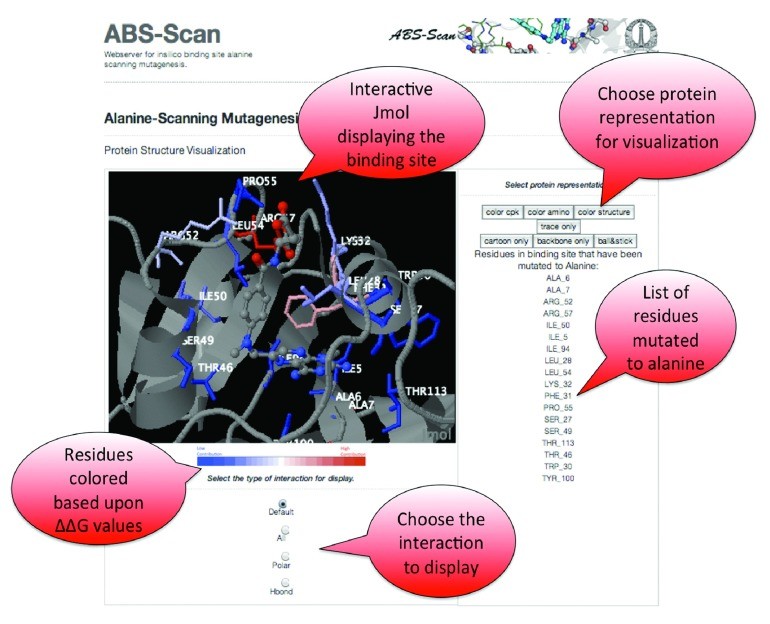
The ABS-Scan web server presents all generated results in an interactive and user-friendly manner directly within the web browser. The Jmol Applet serves as a central visualization tool, displaying the protein-ligand complex and highlighting the contribution of each residue to ligand interaction (Figure 3). Residues are colored using a red-to-blue gradient, with red indicating a higher contribution (larger ΔΔG score) and blue indicating a lower contribution. The Jmol applet also offers options to visualize different types of interactions, such as polar contacts and hydrogen bonds, providing a detailed view of the binding interface.
In addition to the visual representation, ABS-Scan utilizes the d3.js library to generate informative energy plots (Figure 4). These plots display the predicted ΔΔG values for each alanine mutation performed on binding site residues. Residues are ordered on the plot according to their ΔΔG values, allowing for easy identification of the most critical residues. Furthermore, the plots also present the different energy components of the Autodock interaction score for each alanine mutant, providing a breakdown of the energetic contributions. Users can download these plots in publication-quality SVG, PDF, or PNG formats for use in reports and publications.
Beyond the interactive visualization and plots, ABS-Scan provides options to download raw data files. Users can download PDB files of individual mutant structures, enabling further structural analysis. The ΔΔG scores, along with the raw Autodock energy scores, are available for download in CSV format, facilitating data processing and integration with other analysis pipelines. These comprehensive output options ensure that the ABS-Scan alanine scanning tool provides both insightful visualizations and readily accessible data for in-depth analysis of protein-ligand interactions.
{width=828 height=1038}Conclusions
The ABS-Scan web server provides a valuable alanine scanning tool for gaining critical insights into molecular recognition processes involving protein-ligand interactions. Researchers can utilize experimentally determined protein-ligand structures to understand the contribution of individual residues to ligand binding. Furthermore, modeled complexes can be submitted to assess the feasibility of interaction and identify key residues in predicted binding modes. ABS-Scan enhances the analysis of protein binding sites, facilitates comparisons of different ligand interactions, and offers significant utility for researchers conducting alanine scanning mutagenesis studies. We believe ABS-Scan will be a valuable asset to the scientific community, advancing our understanding of protein-ligand interactions and accelerating progress in fields such as drug discovery and protein engineering.
Software Availability
Software access
http://proline.biochem.iisc.ernet.in/abscan/
Latest source code
https://github.com/praveeniisc/ABS-Scan
Source code as at the time of publication
https://github.com/F1000Research/ABS-Scan/releases/tag/V1.0
Archived source code as at the time of publication
http://dx.doi.org/10.5281/zenodo.12806
Software license
ABS-Scan is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Acknowledgements
We gratefully acknowledge all members of the NSC lab for their valuable suggestions during the development of the web-server and result visualization.
Funding Statement
The authors declare that no specific grants were allocated for this project. PA received support from a Bristol-Myers Squibb fellowship during this work.